If your AI product helps diagnose, treat, or guide care, FDA status can change your timeline, study plan, and cost from day one. By early 2026, the FDA had cleared or approved 1,000+ AI/ML medical devices, and about 95% to 97% went through 510(k) rather than De Novo or PMA.
Here’s the short version:
- First, I’d check whether the product is a regulated device at all. Under Section 201(h), AI software can be a medical device. But some software functions may fall outside device rules under Section 520(o).
- Then, I’d match the product to FDA class and risk. Most AI medical devices land in Class II, which often points to 510(k).
- Next, I’d choose the pathway.
- 510(k): used when a predicate exists
- De Novo: used when no predicate exists but risk is lower
- PMA: used for highest-risk products
- After that, I’d build the evidence package. FDA will want data on intended use, model design, datasets, testing, clinical results, cybersecurity, and human factors.
- Last, I’d plan for review and after-market controls. That includes eSTAR, review questions, update limits, and post-clearance monitoring.
A few numbers make the picture clear:
| Pathway | Usual class | Predicate | Typical timing | FDA result |
|---|---|---|---|---|
| 510(k) | Class II | Yes | 3–6 months | Clearance |
| De Novo | Class I/II | No | 6–12 months | Authorization |
| PMA | Class III | No | 12–18+ months | Approval |
A few points stand out:
- Intended use matters more than the AI label
- Black-box clinical tools usually face more scrutiny
- About 50% of AI/ML submissions get an Additional Information request
- A PCCP can help with planned model updates after launch
- As of February 2, 2026, QMSR aligns FDA quality rules with ISO 13485:2016
Put simply, this article explains how I’d go from “Is this even a device?” to “Which FDA path fits?” to “What do I need in the file?” and “What happens after clearance or approval?”
Step 1: Determine Whether Your AI Product Falls Under FDA Oversight
Before you draft a submission, first confirm whether the FDA regulates your product at all. That single call shapes everything that follows: device class, evidence, and the path you can use.
Assess intended use under Section 201(h) and separate regulated from non-device functions
Start with intended use under Section 201(h). Then check whether any function fits the 520(o) software exclusion.
That exclusion applies only if the function:
- analyzes EHR data
- supports, rather than replaces, clinician judgment
- lets the clinician independently review the basis for the recommendation
If a function misses that test, the FDA treats it as regulated. And if you're dealing with a black-box model that a clinician can't independently verify, it will almost never qualify.
Digital Diagnostics' IDx-DR went through De Novo after the FDA found that autonomous diagnosis put it outside the software exclusion [3][7]. That call then shapes what comes next: 510(k), De Novo, or PMA.
If the function is regulated, the next step is to figure out where it lands in FDA classification.
Find the device class, product code, and predicate if one exists
Once you've confirmed a function is regulated, search the FDA Product Classification Database and the AI-enabled device list maintained by the Digital Health Center of Excellence (DHCoE).
These tools help you find the matching 7-digit regulation number and 3-letter product code.
Next, look for a predicate device. That's a legally marketed product with the same intended use and similar tech features. If you find one, you may be able to file a 510(k).
If no predicate exists and the device is new but still moderate risk, De Novo is usually the right path. A successful De Novo creates a new product classification and product code. In plain English, your device can become the predicate for later 510(k) submissions in that same category.
Still stuck after checking the databases? Submit a 513(g) Request for Information to get a formal, binding determination from the FDA.
Match risk level to Class I, II, or III requirements
Device class affects both the amount of evidence you'll need and how long review may take. For SaMD, the FDA also uses the IMDRF risk matrix to score risk based on two things: the importance of the information the software gives and the severity of the patient's condition.
| IMDRF Category | Significance of Info | Condition State | Typical FDA Class |
|---|---|---|---|
| Category I | Inform clinical management | Non-serious | Class I or II |
| Category II | Drive clinical management | Non-serious / Serious | Class II |
| Category III | Treat or diagnose | Serious | Class II |
| Category IV | Treat or diagnose | Critical | Class III |
In practice, most AI-enabled devices land in Class II. That includes triage tools and image segmentation software. Class III is for the highest-risk uses, such as autonomous diagnostics in critical care, and it requires PMA review.
That risk score points you to the submission route in Step 2.
Once class and predicate status are clear, move to the submission pathway.
sbb-itb-535baee
Step 2: Choose the Right FDA Pathway and Submission Strategy
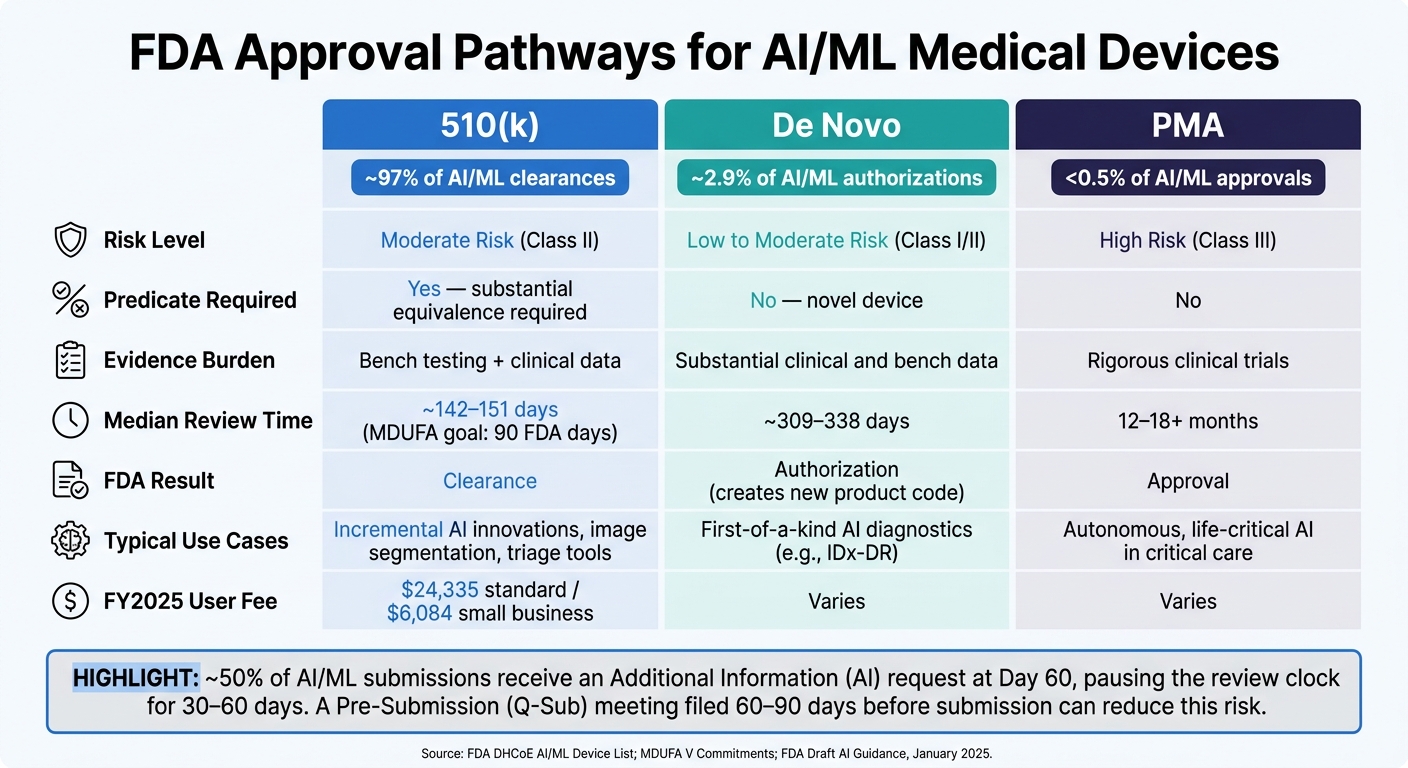
FDA AI Medical Device Pathways: 510(k) vs De Novo vs PMA Compared
Pick the FDA route that matches your device’s class, predicate status, and risk level: 510(k), De Novo, or PMA. That choice shapes the evidence package you’ll build in Step 3.
Compare 510(k), De Novo, and PMA for AI-enabled devices
The right pathway comes down to three factors: risk level, whether a predicate exists, and what the device is meant to do. About 97% of FDA-authorized AI/ML medical devices have cleared through 510(k) [3][7]. Only about 2.9% went through De Novo, and fewer than 0.5% needed PMA [3][7].
That split tells a pretty clear story: most AI devices fall into the moderate-risk Class II bucket and have an available predicate.
| Feature | 510(k) | De Novo | PMA |
|---|---|---|---|
| Risk Level | Moderate (Class II) | Low to Moderate (Class I/II) | High (Class III) |
| Predicate Required | Yes (substantial equivalence) | No (novel) | No |
| Evidence | Bench testing and clinical data | Substantial clinical and bench data | Rigorous clinical trials |
| Median Review Time | ~142 days [3] | ~309–338 days [3] | 12–18+ months [1] |
| Typical Use Cases | Incremental AI innovations | First-of-a-kind AI diagnostics | Autonomous, life-critical AI |
If your product may change after launch, set that boundary now, not later.
Apply FDA Draft AI Guidance and Predetermined Change Control Plans
The January 2025 FDA draft guidance, "Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations," lays out specific content for digital health innovators in AI submissions [8]. It covers device descriptions, user interfaces, data management practices, and model validation documentation.
One of the most useful tools in that guidance is the Predetermined Change Control Plan (PCCP). In plain English, a PCCP lets you spell out in advance which algorithm changes you expect to make after launch and how you’ll validate them, without filing a new 510(k) every time you retrain the model.
A valid PCCP has three parts [5][8]:
- Description of Modifications
- Modification Protocol
- Impact Assessment
In 2025, about 10% of AI clearances included an authorized PCCP [3].
Build the PCCP into the original submission. Trying to bolt it on later is tougher to support and more likely to lead to back-and-forth with FDA reviewers, which can slow down the change envelope you need [7]. This is exactly where a Q-Sub can help. It gives you a chance to pressure-test those assumptions before filing.
Use Pre-Submission Meetings to Reduce Review Delays
A Pre-Submission (Q-Sub) meeting can cut review risk in a very direct way. About 50% of AI/ML SaMD submissions get an "Additional Information" (AI) request around day 60, and that usually stops the review clock for 30–60 days [6]. A solid Q-Sub can lower the odds of that happening.
File your Q-Sub 60–90 days before your planned marketing submission. Keep the discussion centered on the highest-risk issues:
- predicate selection
- clinical study design
- validation methodology
- PCCP scope
After filing, expect about 60–70 days for FDA internal review. Written preliminary feedback usually arrives 2–3 business days before the scheduled meeting [6].
Include a short model card in the Q-Sub package with the model architecture, training data, and key metrics. That gives reviewers a clear reference point [8]. It also makes Step 3 easier, since those answers flow straight into the evidence package.
Step 3: Build an FDA-Ready Evidence Package
Once you’ve picked the pathway in Step 2, the evidence load is mostly set. From there, the job is to build a package that covers the device itself, clinical proof, quality controls, and cybersecurity. Put simply, you need to show what the model does, how it was built, and how you keep it under control.
Define intended use, claims, and a QMSR-ready development process
Your indications for use statement is the base of the whole submission. Every performance claim should tie back to it. Include the intended use, inputs, outputs, workflow, software architecture, UI, level of automation, and model architecture. You also need traceability from claims to validation [8][1].
As of February 2, 2026, QMSR lines up FDA quality requirements with ISO 13485:2016 [2][3]. If your development process didn’t account for that from day one, this is often where costs start to pile up.
Document datasets, validation methods, and clinical performance
After the device is defined, the next step is the data. FDA reviewers want to know where the data came from, how it was collected, how it was cleaned, how it was annotated, and whether it reflects the population the device is meant to serve [8][1].
Your submission should spell out:
- Dataset size
- Collection methods
- Annotation protocols
- Annotator expertise
- How disagreements were resolved
- Any use of synthetic data [8]
Keep the final test set separate from training and tuning [8][2]. That separation matters. If those lines get blurry, performance results can look better than they are.
Performance reporting should include metrics like sensitivity, specificity, and AUC/AUROC. For segmentation tasks, use measures such as Dice Similarity Coefficients and Hausdorff distances [9][6]. FDA also expects subgroup analysis across age, sex, race/ethnicity, plus across clinical sites and equipment types [1][3][9].
A strong clinical package should also show how the AI works in the setting it’s meant for. If the tool supports clinician judgment instead of replacing it, include evidence on human-device team performance [8][9].
Address cybersecurity, bias, and human factors before submission
Close out the premarket file with cybersecurity, bias, and human factors evidence. These aren’t nice-to-have items. They’re part of the submission.
Your file should include an SBOM using CycloneDX or SPDX, along with threat modeling, vulnerability assessments, malformed input testing, penetration testing, and proof of a Secure Product Development Framework (SPDF) [9][1][6]. Non-compliant SBOMs can trigger a Refuse to Accept decision [9].
Human factors testing should look at the risk of clinician overreliance on outputs under realistic use conditions [8][9]. Labeling should also give a clear description of the model’s known limits, plus the sources and demographics of the development and validation data [8].
For healthcare organizations deploying AI devices that handle PHI and clinical workflows, Censinet RiskOps™ can support cybersecurity and third-party risk documentation.
Step 4: Submit, Respond to FDA Review, and Plan Postmarket Controls
Assemble the submission and clear technical screening
Once your evidence package is ready, submit it in the required electronic format. Use eSTAR. 510(k)s have used it since October 1, 2023, and De Novos since October 1, 2025 [10][6]. eSTAR is an interactive template, so it can catch missing fields before you send the file in [6].
A complete submission usually includes:
- Administrative forms
- Device description
- Software documentation
- Algorithm or model summary
- Cybersecurity documentation
- Risk analysis
- Bench, analytical, and clinical performance test results
- Labeling
Software documentation falls into two levels: Basic or Enhanced. Enhanced is required when a software failure could lead to death or serious injury [6][1].
FDA then performs technical screening within 15 days to check eSTAR completeness and confirm fee payment [10].
Manage interactive review timelines and authorization milestones
After screening, FDA starts substantive review. By day 60, you’ll get an email that does one of two things: move the file into interactive review or issue an Additional Information request. About 50% of AI/ML submissions get one [6].
That request pauses the review clock. You then have up to 180 days to respond before FDA treats the file as withdrawn [10].
For 510(k)s, the MDUFA decision goal is 90 FDA days. In practice, the median total time from submission to clearance for AI/ML SaMD through the 510(k) path is 142 to 151 days [6][3]. In 2025, 24% of AI/ML 510(k) submissions cleared in under 90 days [3].
FY2025 user fees are:
- $24,335 for a standard 510(k)
- $6,084 for small businesses [6]
| Review Stage | Timeline | What Happens |
|---|---|---|
| Technical Screening | 15 days | eSTAR completeness check and fee verification [10] |
| Substantive Interaction | 60 days | Interactive review begins or AI Request is issued [10] |
| MDUFA Decision Goal | 90 FDA days | Final 510(k) decision [10][6] |
| AI Request Hold | Up to 180 days | Maximum response window before file withdrawal [10] |
Monitor real-world performance, updates, and ongoing cyber risk
Clearance is not the finish line. FDA uses a lifecycle oversight approach, which means review continues through real-world use long after authorization [11][1].
That means you need to watch whether the device still performs within its cleared conditions of use. It also means tracking shifts tied to new data, changing workflows, or use across different devices or sites [11].
If your submission included a PCCP, that plan can let you pre-approve bounded updates, like retraining on new data, without sending in a new 510(k) or PMA, as long as the change stays inside the authorized limits [2][4][11]. A PCCP must include:
Cybersecurity work also continues after clearance. That includes SBOM maintenance, vulnerability tracking, and patching [1]. Those controls help keep the device inside its authorized risk envelope across the full deployed lifecycle.
For healthcare groups managing AI devices in the field, Censinet RiskOps™ can support ongoing cybersecurity oversight and risk management workflows.
Build postmarket controls into the submission plan from the start.
FAQs
How do I know if my AI software is an FDA-regulated device?
Check the software’s intended use under Section 201(h) of the Federal Food, Drug, and Cosmetic Act. In plain English, FDA usually steps in when the software is meant to diagnose, cure, mitigate, treat, or prevent disease.
That can include Software as a Medical Device (SaMD) and Software in a Medical Device (SiMD). By contrast, software used for billing, admin work, or general wellness usually falls outside FDA regulation.
What makes an AI medical device eligible for 510(k) instead of De Novo or PMA?
An AI medical device can go through 510(k) if the manufacturer can show substantial equivalence to a legally marketed predicate device.
That means the new device needs to have the same intended use and either:
- the same technological characteristics, or
- different characteristics that don’t create new questions about safety or effectiveness
If there isn’t a suitable predicate for a low- to moderate-risk device, the right path is De Novo.
For high-risk devices, manufacturers generally need PMA.
What evidence most often causes FDA review delays for AI device submissions?
Clinical validation studies are often where AI device submissions run into the biggest FDA delays. The agency looks past the final model score and digs into the study itself: how the dataset was gathered, how labels were assigned, and how the statistics were handled.
That’s where many teams get stuck.
Delays usually show up when the submission doesn’t clearly explain:
- where the data came from
- how qualified experts assigned the labels
- whether the study population matches actual clinical use
If those details are thin or hard to follow, the FDA may pause review and ask for more support.
Related Blog Posts
- FDA AI/ML Guidance and Vendor Risk: What Healthcare Organizations Need to Know
- Digital Therapeutics Vendor Risk Management: FDA Regulation and Clinical Evidence
- The Cybersecurity-Clinical Safety Connection: Why Medical Device Security Requires AI
- AI Risks in Medical Device Development: Security Solutions
