Medical device cybersecurity does not end after FDA clearance. In U.S. healthcare, post-market disclosure means you need a clear way to find flaws, assess patient safety risk, decide what to report, tell the right people, and track fixes inside your quality system.
Here’s the short version:
- 24% of healthcare organizations reported attacks aimed at medical devices, and 80% of those said patient care was affected.
- For internet-connected cyber devices, Section 524B requires manufacturers to monitor and address post-market vulnerabilities.
- Under QMSR, cybersecurity now sits inside the quality system, not off to the side.
- Some cases can trigger FDA Part 806, Part 803, and CISA reporting clocks.
- A working program needs a PSIRT, a public disclosure path, steady triage, safety review, tested remediation, and written records tied to CAPA and the SBOM.
- In healthcare, a device workaround can affect care delivery, so clinical input must be part of containment and patching.
If I boil the article down to one point, it’s this: post-market disclosure is a patient-safety and compliance process first, and a technical process second.
What matters most is not just finding a flaw. It’s how you handle it after that:
- who owns the case,
- how fast triage happens,
- how clinical risk is judged,
- what gets disclosed,
- and how each case feeds future product and vendor decisions.
A simple way to think about it:
| Area | What it needs to cover |
|---|---|
| Governance | PSIRT roles, escalation path, executive sign-off |
| Intake | Public reporting channel, safe-harbor language, case logging |
| Triage | Validation, CVSS v4.0 scoring, SBOM review, safety check |
| Response | Interim controls, verified fix, customer guidance |
| Disclosure | Advisory content, FDA/CISA notices where required |
| Follow-up | CAPA linkage, SBOM updates, portfolio tracking |
So if you’re a manufacturer or an HDO, the goal is simple: make disclosure repeatable, documented, and safe for patient care.
Cybersecurity in Medical Devices – What QA/RA Must Do Today
sbb-itb-535baee
The U.S. Regulatory and Industry Framework for Disclosure
Once a vulnerability is found, disclosure doesn't happen in a vacuum. In the U.S., it sits inside FDA reporting rules, coordinated vulnerability handling, and cyber incident response requirements.
FDA Postmarket Cybersecurity Expectations

Postmarket cybersecurity is now part of the regulatory and quality system framework. Section 524B of the FD&C Act requires manufacturers of internet-connected "cyber devices" to monitor, identify, and address postmarket vulnerabilities promptly [1]. Under QMSR, cybersecurity sits inside design controls and CAPA [1][2].
That means manufacturers can't treat security issues as side work. They need a steady process for watching sources like the NVD, ICS-CERT advisories, vendor bulletins, and risks tied to components listed in the SBOM. From there, teams should score findings with CVSS v4.0 and confirm clinical impact under ISO 14971 [1].
Reporting duties depend on what happened and how much harm is involved:
- Report corrective actions that reduce a health risk under 21 CFR Part 806 within 10 working days [1].
- Report death or serious injury under 21 CFR Part 803 [1].
- Under CIRCIA, report covered cyber incidents to CISA within 72 hours and ransomware payments within 24 hours [2].
Coordinated Vulnerability Disclosure and Sector Coordination
CVD works best when researchers know where to report an issue and what happens next. That starts with a public intake path and safe-harbor language that protects good-faith researchers who stay within defined scopes and reporting behaviors [1].
The usual target is a 90-day public advisory timeline [1]. If a fix touches a safety-critical issue and needs more time, silence is a bad move. Keep the reporter and other stakeholders updated so the process doesn't stall.
Things get tougher when a flaw affects shared components used by multiple vendors. In those cases, CISA's coordination process helps line up disclosure across parties, and the Traffic Light Protocol (TLP) helps control who can see what information. For example, TLP:AMBER supports limited distribution [1].
Health-ISAC adds another layer by helping with threat sharing. And the HSCC MedTech Vulnerability Communications Toolkit gives manufacturers, HDOs, and regulators a common way to talk during a disclosure event [2]. That's more than paperwork. It cuts confusion when timing and wording matter.
How NIST and Healthcare Playbooks Support Incident Handling
NIST and healthcare playbooks turn reporting rules into repeatable response steps. NIST SP 800-61 lays out the base structure in four phases: preparation, detection and analysis, containment and eradication, and post-incident activity [2].
Two healthcare-focused playbooks build on that structure and make it usable in clinical settings:
- The FDA Medical Device Cybersecurity Regional Incident Preparedness and Response Playbook, developed with MITRE, focuses on HDO coordination [2].
- The Medical Product Manufacturer Cyber Incident Response Playbook (HSCC, 2024) covers manufacturer duties across the device lifecycle [2].
Together, these playbooks adapt NIST to clinical environments [2].
A practical piece inside this setup is a three-tier classification system. It helps teams decide how fast to move and how far to spread the response. The table below shows how those tiers map to real situations [2]:
| Tier | Classification | Example |
|---|---|---|
| Tier 1 | Critical - active exploitation with probable patient safety impact | Ransomware encrypting drug delivery controls |
| Tier 2 | Major - confirmed compromise with potential safety impact | Unauthorized access to device configuration |
| Tier 3 | Minor - suspected compromise of supporting or administrative systems with no direct patient safety impact | Phishing attack on administrative support staff |
Every decision should be documented. Teams should record patient-safety assessments, CAPA records, design changes, SBOM revisions, and the basis for each reporting decision [1][2]. Even when a vulnerability is treated as routine and not reportable under Part 806, the written clinical impact rationale can matter later during an audit.
Those tiers and records shape the response and communication steps that come next.
Core Elements of a Post-Market Cybersecurity Disclosure Program
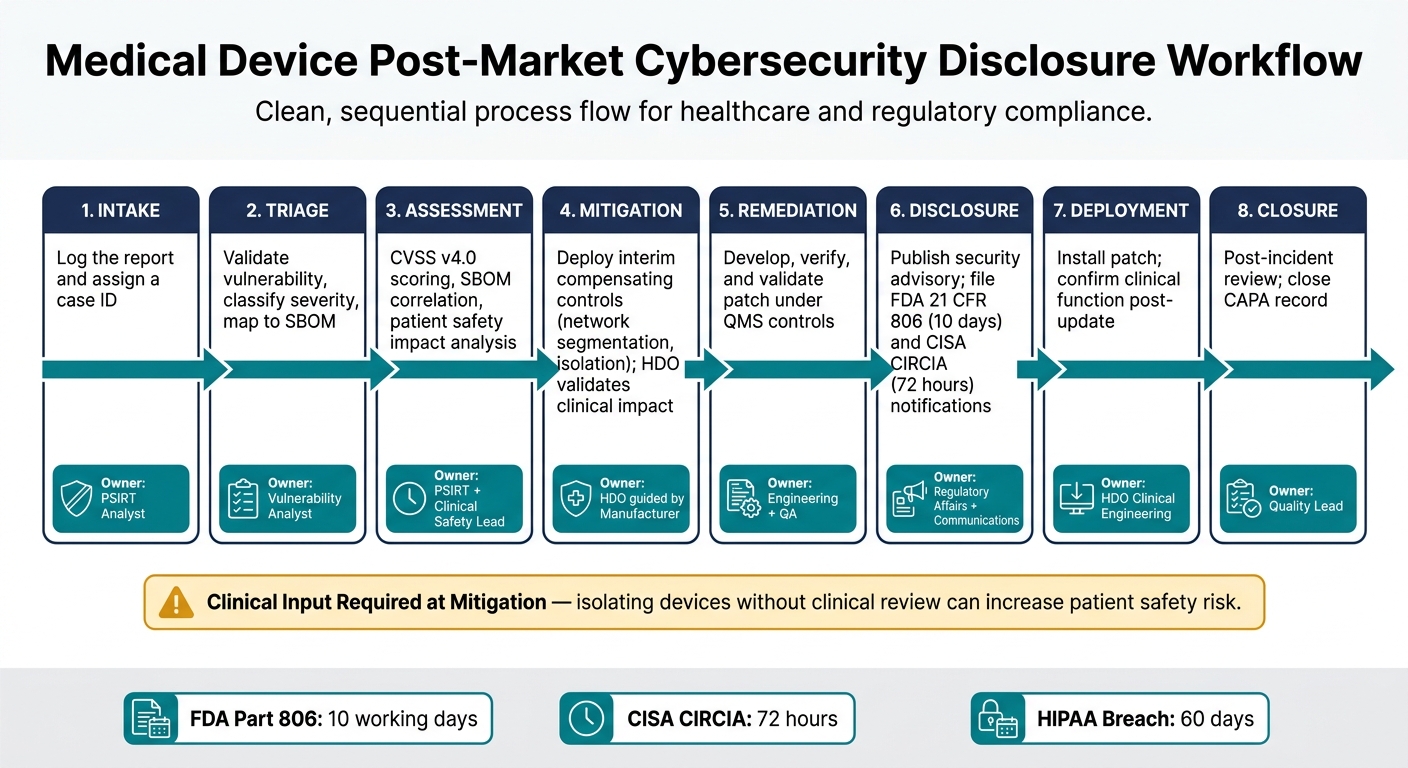
Medical Device Post-Market Cybersecurity Disclosure Workflow
A disclosure program works only when the structure is clear. If roles are fuzzy, processes aren't written down, or scoring shifts from case to case, teams end up making rushed calls under pressure. That's usually when the cost of a mistake goes up fast. The point is simple: turn vulnerability reports into action that protects patients and stands up to regulator review. That starts with clear ownership, so intake and triage don't stall.
Governance, Roles, and Decision Authority
At the center of the program is a Product Security Incident Response Team (PSIRT). This is the formal group that receives, assesses, and resolves product security vulnerabilities, so manufacturers can coordinate with researchers, HDOs, and regulators [1]. Under Section 524B of the FD&C Act, manufacturers of "cyber devices" are legally required to maintain a coordinated vulnerability disclosure (CVD) plan and the procedures that support it [1].
Each PSIRT role should have one clear owner:
| Role | Primary Responsibility |
|---|---|
| PSIRT Lead | Overall coordination, executive escalation, and final notification decisions |
| Vulnerability Analyst | Triage, CVSS v4.0 scoring, and SBOM correlation |
| Clinical Safety Lead | Assessing patient harm potential under ISO 14971 |
| Regulatory Affairs | Managing FDA 21 CFR Part 806 and CISA notifications |
| Communications | Drafting public security advisories and coordinating with external researchers |
Under the FDA's QMSR, PSIRT work sits inside the quality system and must feed design controls and CAPA [1]. That means this isn't just an IT function off to the side. It has to connect back to quality, risk, and product change control. Executive sponsors also need visibility for resource calls and final sign-off on regulatory submissions.
Once ownership is clear, the team can move from intake to disclosure decisions without wasting time on internal confusion.
Vulnerability Intake, Triage, and Risk Scoring
A working intake process begins with public reporting channels: a dedicated security email such as security@company.com, a structured web form, and PGP keys for encrypted submissions [1]. The program also needs safe harbor language. That language protects good-faith researchers from legal action as long as they stay within the stated scope and behavior rules [1].
After a report arrives, timing matters. The team should acknowledge it within 48 hours and finish initial triage within 5 business days [1]. Triage includes validating the report, scoring it with CVSS v4.0, and checking the affected component against the SBOM to see which devices may be exposed [1].
Prioritization should follow one steady framework tied to exploitability and clinical impact:
| Priority | Criteria | Response Target |
|---|---|---|
| Critical | Active exploitation + direct patient safety risk | 24–72 hours |
| High | Exploitable and safety-relevant | 1–2 weeks |
| Medium | Exploitable, no direct safety impact | 30–60 days |
| Low | Theoretical, significant barriers to exploitation | 90 days |
Every triage decision should be recorded in the QMS. FDA inspectors review vulnerability handling and PSIRT records during QMS audits [1]. If the recordkeeping is weak, even a sound technical response can become hard to defend.
Program Maturity Benchmarks
These controls also show whether a program can measure what matters for cybersecurity and keep working when pressure hits. The table below shows the difference between a bare-bones setup and one that can hold up during regulator review and coordination across organizations [1]:
| Feature | Minimal Program | Intermediate Program | Mature Program |
|---|---|---|---|
| Policy Coverage | Ad hoc response; no public policy | Public disclosure policy; defined scope | Comprehensive policy with safe harbor and SLAs |
| Monitoring | Reactive (customer reports only) | Periodic NVD/CISA KEV checks | Real-time telemetry; proactive threat hunting |
| Response Timeline | Undefined; often >120 days | 90-day standard disclosure window | Accelerated timelines (24–72 hours for critical) |
| Stakeholder Coordination | Internal only | Ad hoc ISAC sharing | Active H-ISAC/CISA coordination with TLP usage |
| Documentation Quality | Basic email logs | Structured incident IDs; CAPA linkage | Full QMS integration; SBOM-to-vulnerability mapping |
| Remediation Tracking | Manual patches | Validated updates; customer advisories | Automated deployment; verified clinical workarounds |
The gap here is both operational and regulatory. Mature programs close that gap with continuous monitoring, structured coordination, and documentation that is ready for audit from day one.
How to Coordinate Incident Response and Stakeholder Communication
A mature disclosure program spells out what happens next, who owns it, and where decisions get made for every report. Once those roles are set, the work moves into execution: intake, triage, mitigation, and disclosure.
From Intake to Remediation
The workflow below reflects the response process established in the previous section [1]. Each phase has a clear owner, which helps the manufacturer and the HDO stay in sync as the case moves forward.
| Phase | Action | Owner |
|---|---|---|
| 1. Intake | Log the report and assign an ID | PSIRT Analyst |
| 2. Triage | Validate the vulnerability, classify it, and map affected components to the SBOM | Vulnerability Analyst |
| 3. Assessment | Complete CVSS v4.0 scoring, SBOM correlation, and patient safety impact analysis | PSIRT + Clinical Safety Lead |
| 4. Mitigation | Deploy interim compensating controls, such as network segmentation or selective isolation - manufacturer provides guidance; HDO validates clinical impact | HDO, guided by the manufacturer |
| 5. Remediation | Develop, verify, and validate the patch under QMS controls | Engineering + QA |
| 6. Disclosure | Publish the security advisory and file required FDA and CISA notifications | Regulatory Affairs + Communications |
| 7. Deployment | Install the patch and confirm clinical function after update - manufacturer provides the fix; HDO validates clinical impact | HDO Clinical Engineering |
| 8. Closure | Complete the post-incident review and close the CAPA record | Quality Lead |
Phase 4 needs special care. Containment can’t be treated like an IT-only move when patient care is on the line. The FDA/MITRE playbook says it plainly:
"A purely technical decision to isolate devices without clinical input can create greater patient safety risk than the cyber incident itself." [2]
Once containment begins, the advisory needs to give HDOs clear steps they can act on right away.
What Disclosure Messages Should Contain
A disclosure message should tell HDOs exactly how to assess, mitigate, and patch the issue. If the message is vague, HDO teams end up chasing answers instead of fixing the problem.
Each disclosure message should include:
- Affected products: Full product name, model number, software version, and UDI-DI/PI where applicable
- Vulnerability description: CVE identifier, CVSS v4.0 score, and a plain-language summary
- Exploit conditions: What an attacker would need - such as network access, authentication, or physical proximity - to exploit the issue
- Clinical impact: A clear statement of whether patient safety or clinical workflows could be affected
- Interim mitigations: Specific compensating controls HDOs can apply at once, such as network segmentation, selective isolation, or manual clinical workarounds
- Permanent fix: The exact software version that resolves the issue, with step-by-step installation instructions
- Support contact: A named team or email address for follow-up questions
- Update cadence: Next update date
Regulatory notice timelines run alongside customer communications. Manufacturers must file an FDA 21 CFR 806 report within 10 working days if a correction starts to reduce a risk to health, notify CISA within 72 hours of a covered cyber incident under CIRCIA, and report a HIPAA breach within 60 days if unsecured PHI is involved [2].
Response Ownership by Scenario
The right coordination model depends on where critical medical device security risks show up. Sometimes the manufacturer finds it first. Other times it surfaces inside an HDO’s environment. In some cases, it’s already being used across multiple sites, which changes the pace and the communication model.
| Feature | Manufacturer-Led | HDO-Led | Jointly Coordinated |
|---|---|---|---|
| Primary Owner | Manufacturer PSIRT | HDO Security/Clinical Engineering | PSIRT + HDO Risk Management |
| Disclosure Lead | Manufacturer Product Manager | HDO IT/Internal Comms | Joint Advisory (Manufacturer + CISA/H-ISAC) |
| Technical Validation | Internal QA/Regression Testing | HDO Sandbox/Clinical Test Environment | Manufacturer validates fix; HDO validates workflow impact |
| Regulatory Touchpoints | Documented in QMS/Vulnerability Plan | Internal HIPAA/Compliance records | FDA 524B/MDR Reporting; CISA Coordination |
| Timeline Pressure | Standard 90-day disclosure cycle | Immediate containment (hours to days) | Accelerated: 24–72 hours for critical safety risks |
Joint coordination works best when status updates are shared and one disclosure lead is clearly named. Active participation in H-ISAC using Traffic Light Protocol (TLP) labels - TLP:AMBER for limited distribution and TLP:GREEN for community-wide sharing - helps keep sensitive details with the right audience while still supporting sector-wide awareness [1]. That shared picture is what allows portfolio-wide tracking.
For HDOs managing large device portfolios, Censinet RiskOps™ can centralize affected-device status, vendor responses, and remediation tracking.
Those disclosure records should then feed into enterprise risk management and continuous improvement.
Integrating Disclosure into Enterprise Risk Management and Continuous Improvement
Using Disclosure Data to Improve Future Security and Compliance
Once a disclosure closes, the work shouldn't stop with the case file. The findings need to feed into enterprise risk. Each disclosure should connect back to CAPA, design controls, and procurement criteria. Under QMSR, post-market findings must map to ISO 13485 Clause 7.3 and Clause 8.5 [1]. If those findings aren't mapped, security gaps and compliance gaps can slip through.
Recurring vulnerabilities matter even more. If the same issue shows up in one third-party component again and again, or appears across several device families, that should trigger CAPA. In many cases, that points to a deeper procurement or architecture problem, not a one-off defect.
Vendor security data should also shape buying decisions. The market is already moving that way:
- 35% of healthcare procurement decision-makers will no longer evaluate a device that lacks an SBOM [3]
- 81% rate SBOMs as important or essential [1]
Teams should also track time-to-triage and time-to-remediate against fixed SLAs [3].
Those records shouldn't live in isolation, either. They should feed portfolio-wide risk visibility so teams can spot patterns, compare vendor performance, and see where remediation is stalling.
Centralizing Risk Visibility Across Devices, Vendors, and Facilities
Most HDOs need one clear view across vendors, facilities, and device types. Device portfolios often include dozens of vendors, multiple facilities, and a mix of legacy and modern systems. Without a central view, remediation tracking gets fragmented fast, and executive reporting starts to lose accuracy.
The numbers show why this matters. 44% of healthcare organizations acknowledge running devices with known, unpatched vulnerabilities, and 28% operate devices past their end-of-support date [1]. When that's the baseline, centralized remediation tracking isn't optional.
Centralized reporting keeps disclosure data visible across vendors, devices, and facilities. Censinet RiskOps™ gives HDOs and vendors a structured way to manage that sprawl, centralizing third-party and enterprise risk workflows and giving risk teams command center visibility.
Conclusion: Key Practices Every Disclosure Program Should Have
A strong post-market cybersecurity disclosure program comes from consistent habits across governance, operations, and continuous improvement. The core practices are:
- Align with FDA expectations under Section 524B and the 2026 QMSR update, treating cybersecurity as a quality system obligation, not a standalone IT function [1][2]
- Formalize coordinated vulnerability disclosure with a documented CVD policy, a functioning PSIRT, and active H-ISAC/CISA coordination [2]
- Define response ownership clearly so every incident has a named lead and a known escalation path
- Standardize disclosure documentation so each case captures technical severity, clinical impact, and response timelines
- Preserve evidence through CAPA records and SBOM updates that connect each disclosure back to design controls and procurement criteria
Each disclosure should update controls, procurement, and remediation priorities so the same issue doesn't keep coming back. Teams that tie each case to enterprise risk management - by updating inventories, tightening vendor contracts, and reporting measurable progress to leadership - are in a much better position to protect patients and the organization over time.
FAQs
When does a cybersecurity issue become reportable?
A cybersecurity issue becomes reportable when it hits a regulatory trigger based on what happened and how much it affected safety or operations.
- Under 21 CFR Part 806, report corrections made to reduce a health risk that could cause death or serious injury within 10 working days.
- Under 21 CFR Part 803, report actual death, serious injury, or device malfunction through the eMDR system.
- Under CIRCIA, covered entities must report covered incidents to CISA within 72 hours and ransomware payments within 24 hours.
Who should own post-market vulnerability disclosure?
A dedicated Product Security Incident Response Team (PSIRT) should handle post-market vulnerability disclosure. This team receives, reviews, and resolves vulnerabilities throughout a device’s commercial life.
The PSIRT Lead manages coordination, escalation, and regulatory notification. They’re backed by vulnerability analysts, security engineers, regulatory affairs, and product management.
How do manufacturers balance patching with patient safety?
Manufacturers have to balance patching with patient safety. That’s why many fold cybersecurity into clinical risk management and use ISO 14971 to judge how a vulnerability could affect a device’s safety and essential performance.
They don’t stop at technical severity scores like CVSS. They also look at possible patient harm and any compensating controls already in place. If a vulnerability is critical, it may call for an immediate, out-of-cycle patch. More routine issues are usually handled through regular update cycles, with clear justification for the timing.
